Artificial intelligence helps bring about record-fast vaccines
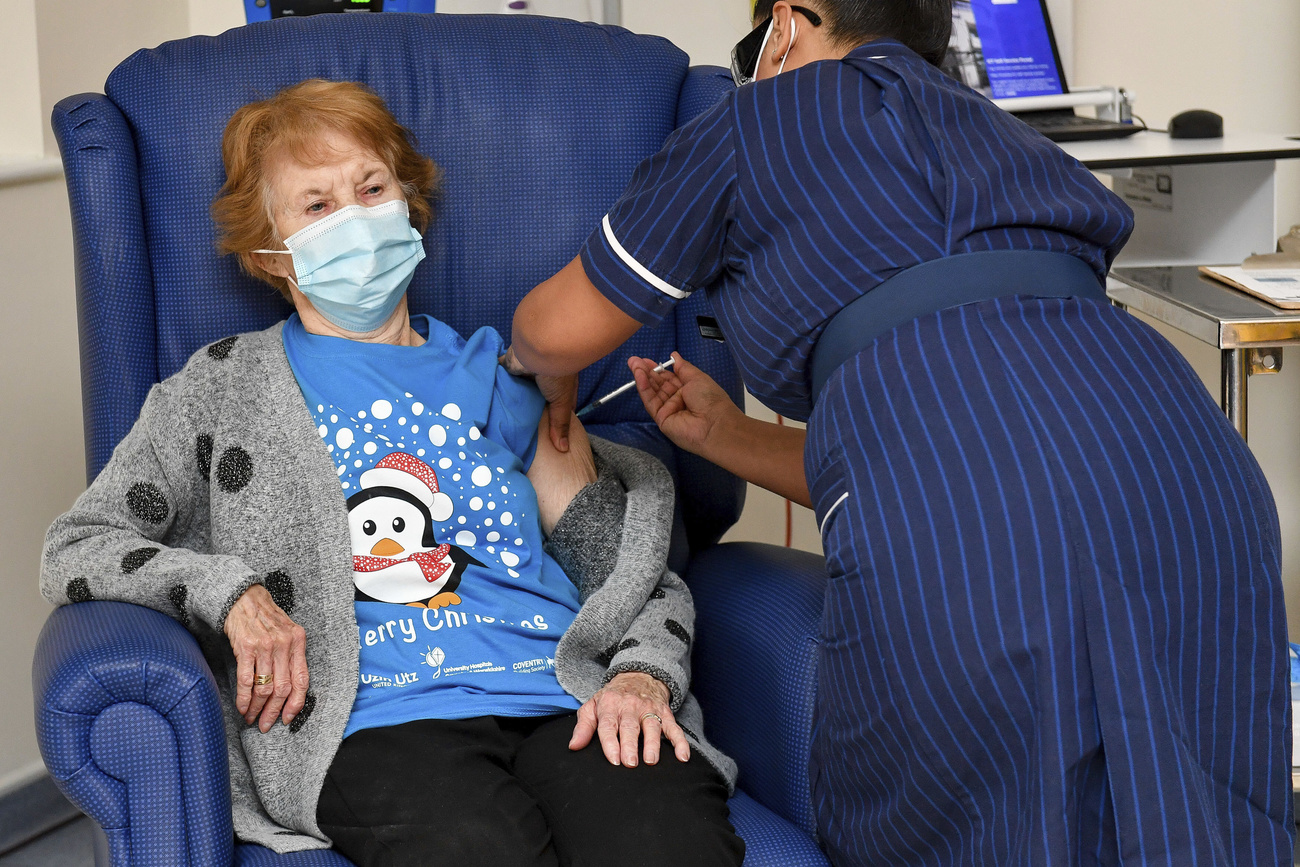
The rapid development of two highly effective Covid-19 vaccines was made possible through AI technology and innovative collaboration among researchers around the world, including Switzerland.

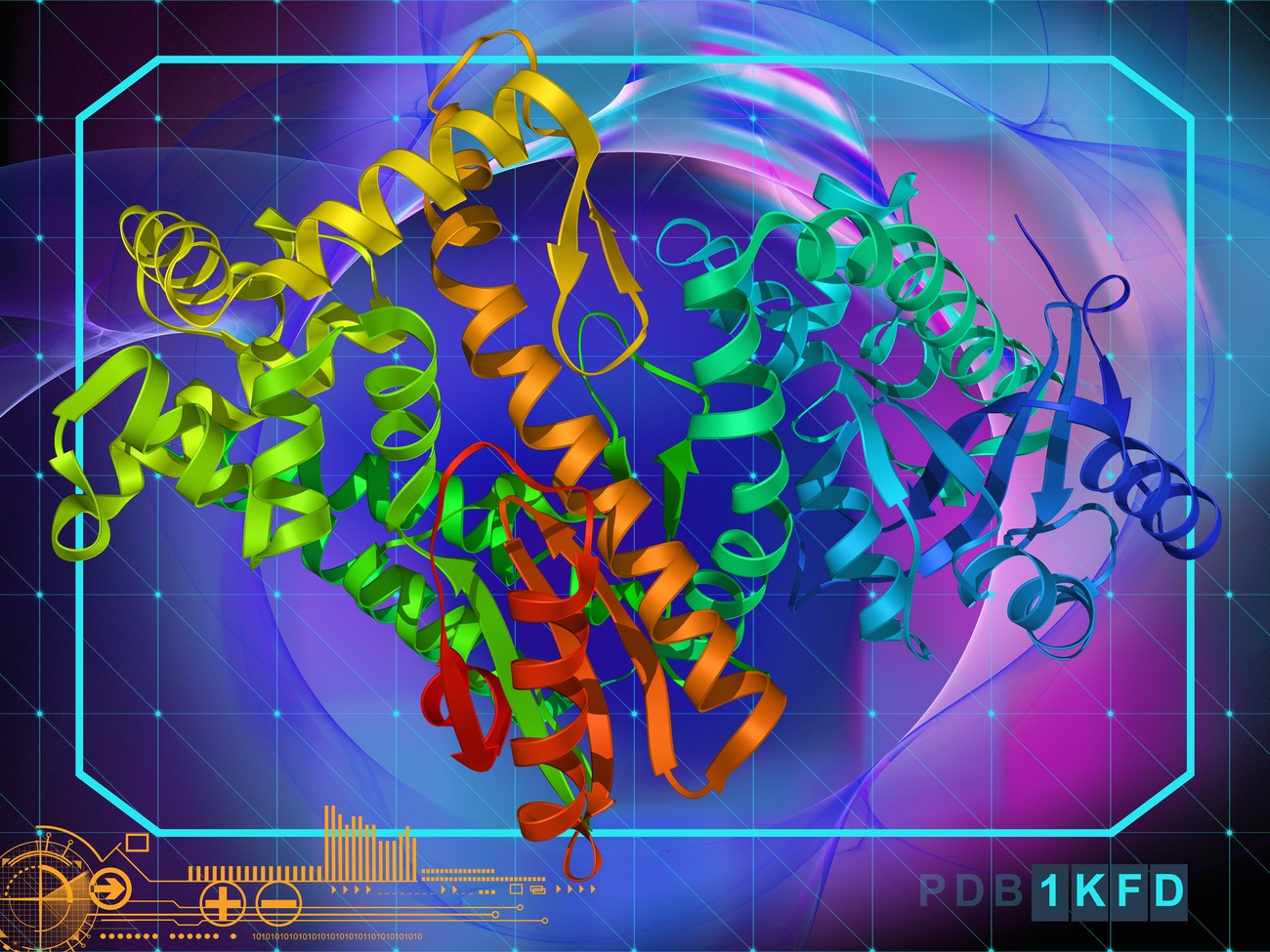
Artificial intelligence (AI) has the potential to revolutionise the world of science and solve some of the most complex problems facing modern biology. Chief among them: predicting the structure of unknown proteins to unlock the secrets of cells and the diseases that affect them. Recently, protein structures have been in the spotlight because of their central role in the development of new Covid-19 vaccines, such as those based on messenger RNA.
Solving protein structures experimentally is a long and laborious process, requiring months of research and lots of resources. Structural information is essential for studying new viruses, understanding their behaviour, and developing effective vaccines. Scientists’ newfound ability to predict proteins’ structures through computational methods has made this process much faster and much more accurate.
Thanks to recent advances in artificial intelligence, it is now possible to predict the three-dimensional structures of very challenging target proteins with high accuracy. A milestone was achieved when AlphaFold2, the AI system created by London-based company DeepMind, made it possible to rapidly determine several protein structures of SARS-CoV-2, a virus about which we knew very little until a few months ago. (DeepMind has been owned by Google since 2014). Scientists’ relentless work and international collaboration – with the help of cutting-edge AI technologies like DeepMind – made it possible to respond quickly to the pandemic. Currently, as many as 60 potential vaccines are in various stages of clinical development and three have been approved for widespread use by certain national regulatory authorities, according to the World Health OrganizationExternal link.
Biomedical researchers consider this moment a turning point for science. ”It’s an absolutely incredible achievement,” says Torsten Schwede, vice president for research at the University of Basel and head of the research group at the SIB Swiss Bioinformatics Institute, which develops SWISS-MODEL, a fully automated protein structure modelling server used by researchers worldwide. DeepMind’s achievements were also made possible by the progress of the last ten years in the field of computational structural biology, of which the SWISS-MODEL system was a pioneer.
People and software
Why are proteins taking centre stage in the medical and scientific field? Small but critically important, proteins underlie the chemical and biological processes of human cells and any living organism. The amino acids that compose them bind together, forming spontaneous “origami”, which determines the unique three-dimensional structure of proteins. Knowing their shape greatly facilitates biomedical research, for example in the field of human diseases, and it’s the reason why the scientific community considers the results achieved by DeepMind revolutionary. The hope is that this achievement will translate into the development of new medicines and advanced treatments.
SWISS-MODEL was the world’s first automated software capable of autonomously modelling the three-dimensional structure of proteins that had not yet been discovered through experiments. In 1993, Manuel Peitsch, bioinformatician and founder of SWISS-MODEL, launched the idea of using computer simulations that did not require human intervention to get structural information about proteins and better understand molecular functions.
At the time, this project seemed like science fiction. Today, thanks to increasingly sophisticated methods of comparing known protein structures with sequences of unknown proteins – so-called ”homology modelling” – the software has surpassed human capabilities in terms of accuracy and performance and is used worldwide. Each year, SWISS-MODEL processes more than one million requests for protein models without human supervision.
Proteins are flexible objects which often need to move in order to function. To compare a 3D model of a protein with an experimental reference structure, the models must be “rotated” until there is an optimal overlap. But for flexible objects, this superposition is difficult to achieve. To solve this problem, the SWISS-MODEL team developed a score called LDDT (Local Distance Difference Test), which evaluates and predicts to what degree a protein will overlap with the model structure, regardless of its movements. Such scores, which can be used without human supervision, are essential for the development of autonomous protein modeling methods.
Reality and science fiction
For the past three decades, scientists have been trying to extract the characteristic three-dimensional structures of proteins from their amino acid sequences. In this process, knowing experimental structures of related proteins makes modeling relatively easy and accurate. But in difficult cases, the absence of structural information within a protein family means making a very complicated and often inaccurate prediction from scratch. But thanks to AlphaFold2, that problem no longer exists.
”We saw that the AlphaFold2 method developed by DeepMind works for both easy and very difficult cases. It’s a real breakthrough, because now AI can accomplish something that no human with in-depth knowledge of protein modeling had been able to do before,” Schwede says.
DeepMind’s AI system, AlphaFold2, uses advanced machine learning techniques, known as deep neural networks, to predict protein structures directly from their genetic sequences. To do so, the AI system learned the sequences and structures of about 100,000 known proteins by making use of experimental data made available by the scientific community. It can now make highly accurate 3D model predictions for any protein. DeepMind’s extraordinary achievements were confirmed by the organisers of the CASP experiment on protein structuresExternal link (see box), who called AlphaFold2’s calculation and prediction “unprecedented”.
CASP (Critical Assessment of protein Structure Prediction) is an experiment that takes place every two years and evaluates developments in the field of protein structure prediction at the international level. During the recent experiment held in 2020 (CASP14), the accuracy of the prediction methods was evaluated on nearly 100 protein targets. AlphaFold2’s predictions were shown to be very accurate, even in difficult cases, such as a previously unknown SARS-CoV-2 protein, ORF8. SARS-CoV-2 is composed of about 30 different proteins, a dozen of which are poorly understood.
The golden rule of progress
The strong support of the scientific community and the open exchange of information in the field of computational structural biology, to which SWISS-MODEL has also contributed, are some of the explanations for DeepMind’s success. According to Torsten Schwede, the open sharing of computational methods and structural data gave DeepMind the information it needed to solve one of the most challenging problems in bioinformatics.
The exchange intensified during the pandemic, demonstrating the importance of collaboration to achieve meaningful results in significantly shorter time frames.
”We learned a lot during this pandemic. The open sharing of information on SARS-CoV-2 within the scientific community made it possible to develop vaccines in record time,” Schwede adds, noting that this proves how counterproductive data silos can be to scientific achievement.

More
Aussie adoptee gains Swiss citizenship at 54 thanks to old envelope

More
Thousands of emigrants’ descendants in Argentina demand Swiss citizenship

More
Switzerland’s most renowned trans person no longer wants to be a woman

More
Solothurn, the Swiss city that goes up to 11

More
Explainer: Switzerland gears up for driverless transport era




In compliance with the JTI standards
More: SWI swissinfo.ch certified by the Journalism Trust Initiative



Join the conversation!