Vacunas en tiempo récord: la inteligencia artificial hace su contribución a la ciencia
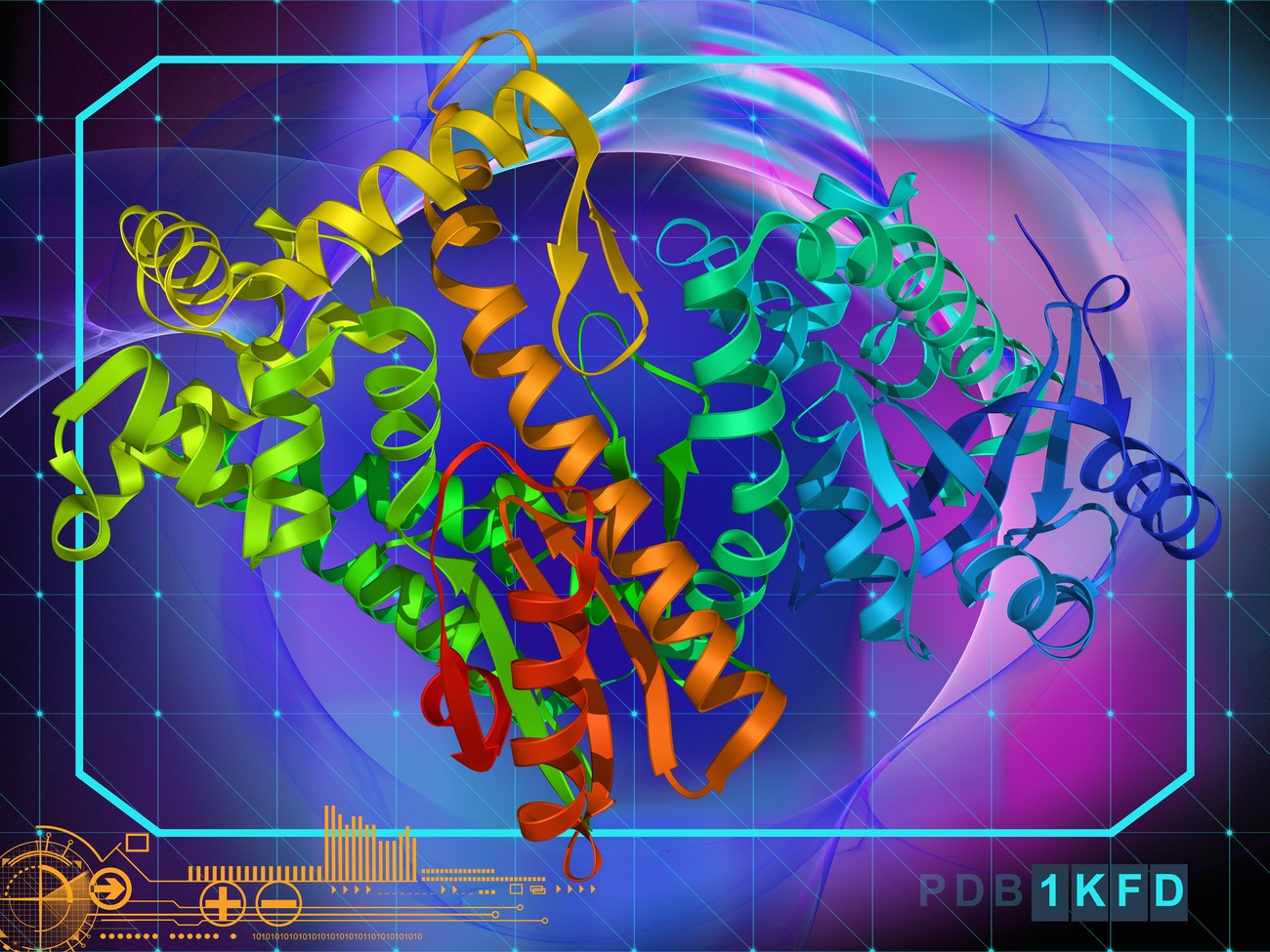
El rápido desarrollo de dos vacunas contra la COVID-19 altamente efectivas ha sido posible gracias al desarrollo tecnológico de la inteligencia artificial y a la colaboración innovadora entre investigadores de todo el mundo, incluida Suiza.

Detrás de los desafíos éticos asociados a la inteligencia artificial (IA) se esconden las enormes posibilidades de una tecnología que podría revolucionar el mundo de la ciencia y resolver algunos de los problemas más complejos de la biología moderna. En primer lugar figura la capacidad de predecir la estructura de proteínas desconocidas para revelar los secretos de las células y las enfermedades que las afectan. Recientemente, las estructuras de proteínas han sido noticia debido a su papel central en el desarrollo de nuevas vacunas contra la COVID-19, vacunas basadas en el ARN mensajero.
Definir la forma de las proteínas de forma experimental es un proceso largo y laborioso, que requiere meses de investigación y un elevado consumo de recursos. Esa información es esencial para estudiar los nuevos virus, comprender su comportamiento y desarrollar vacunas eficaces. La capacidad desarrollada por los científicos para predecir las estructuras de las proteínas mediante métodos de cálculo informatizados ha hecho que ese proceso sea mucho más rápido y preciso.
Gracias a los recientes avances en inteligencia artificial, es posible ahora predecir con gran precisión las estructuras tridimensionales de proteínas diana de alta complejidad. Se dio un paso muy importante cuando AlphaFold2, el sistema de IA creado por la empresa londinense DeepMind (propiedad de Google desde 2014), permitió definir rápidamente diferentes estructuras proteínicas del SARS-CoV-2, un virus sobre el que se disponía de muy poca información hasta hace solo unos meses.
El trabajo incansable de los científicos y la colaboración internacional -con la ayuda de tecnologías de IA de vanguardia como DeepMind- han hecho posible reaccionar rápidamente ante la pandemia. Actualmente, se encuentran en desarrollo clínico hasta 60 posibles vacunas, tres de las cuales han sido ya aprobadas por algunas autoridades reguladoras nacionales para su utilización a gran escala, según indica la Organización Mundial de la SaludEnlace externo.
Los investigadores del mundo de la biomedicina ven este momento como un punto de inflexión para la ciencia. «Es un logro absolutamente increíble», comenta Torsten Schwede, vicepresidente de investigación de la Universidad de Basilea y jefe del grupo de investigación del Instituto Suizo de Bioinformática SIB, que ha desarrollado SWISS-MODEL, un servidor de modelización de estructuras de proteínas totalmente automatizado y utilizado por investigadores de todo el mundo. Los resultados obtenidos por DeepMind también fueron posibles gracias a los avances de los últimos diez años en el campo de la biología estructural computacional, del que el sistema suizo SWISS-MODEL fue pionero.
«El intercambio abierto de información sobre el SARS-CoV-2 en el seno de la comunidad científica ha hecho posible desarrollar vacunas en un tiempo récord».
Torsten Schwede
Personas y programas informáticos
¿Por qué las proteínas ocupan un lugar tan destacado en el campo médico-científico? Pequeñas, pero de importancia fundamental, las proteínas son la base de todos los procesos químicos y biológicos de las células humanas y de cualquier organismo vivo. Los aminoácidos que las componen se unen entre sí formando origamis espontáneos, que determinan su particular estructura tridimensional.
Conocer la forma de las proteínas facilita enormemente la investigación biomédica en el terreno de las enfermedades humanas, por ejemplo. Por eso, la comunidad científica considera revolucionarios los resultados obtenidos por DeepMind. Hay una gran esperanza en que este logro se traduzca en el desarrollo de nuevos medicamentos y tratamientos farmacológicos avanzados.
SWISS-MODEL creó el primer software del mundo capaz de modelar de forma completamente autónoma las estructuras tridimensionales de proteínas aún no descubiertas por método experimental. En 1993, Manuel Peitsch, bioinformático y fundador de SWISS-MODEL, lanzó la idea de utilizar sistemas de simulación por ordenador que no requirieran intervención humana para obtener información estructural sobre proteínas y comprender mejor las funciones moleculares.
En aquel momento el proyecto parecía una cosa de ciencia ficción. Hoy en día, gracias a los métodos cada vez más sofisticados para comparar estructuras proteínicas conocidas con secuencias de proteínas desconocidas -lo que se conoce como “modelización por homología”- los software han superado la capacidad humana en términos de precisión y rendimiento y se utilizan en todo el mundo. Cada año, SWISS-MODEL procesa más de un millón de solicitudes de modelos de proteínas sin supervisión humana.
Las proteínas son objetos flexibles. Para que funcionen necesitan a menudo movimiento. Para comparar un modelo 3D de una proteína con una estructura de referencia experimental, hay que hacer rotar el modelo hasta que haya una superposición óptima. Pero con los objetos flexibles, esta superposición es difícil de lograr. Para resolver este problema, el equipo de SWISS-MODEL desarrolló un marcador llamado lDDT (local Distance Difference Test), que evalúa el grado de concordancia de una predicción con respecto a la estructura de referencia, independientemente de los movimientos intramoleculares. Esos marcadores, que pueden utilizarse sin necesidad de supervisión humana, son esenciales para el desarrollo de métodos autónomos de modelado de proteínas.
Realidad y ciencia ficción
Durante los últimos treinta años, los científicos han intentado deducir la característica forma tridimensional de las proteínas a partir de sus secuencias de aminoácidos. En ese proceso, conocer las estructuras experimentales de las proteínas emparentadas entre sí hace que el modelado sea relativamente fácil y preciso. En los casos difíciles, sin embargo, la ausencia de información estructural sobre una familia determinada de proteínas significa hacer una predicción ex novo muy complicada y, a menudo, inexacta. Pero gracias a AlphaFold2, este problema se ha resuelto. «Hemos visto que el método AlphaFold2 desarrollado por DeepMind funciona tanto para casos fáciles como muy complejos. Es un auténtico avance, porque ahora la IA puede lograr algo que ningún ser humano con un conocimiento profundo del modelado de proteínas había sido capaz de hacer antes «, afirma Schwede.
El sistema de inteligencia artificial de DeepMind, AlphaFold2, utiliza técnicas avanzadas de aprendizaje automático, conocidas como redes neuronales profundas, para predecir las estructuras de las proteínas directamente a partir de sus secuencias genéticas. Para ello, el sistema de IA aprendió las secuencias y estructuras de unas 100 000 proteínas conocidas utilizando datos experimentales facilitados por la comunidad científica. Hoy, es capaz de realizar predicciones muy precisas de modelos 3D de cualquier proteína. Los extraordinarios resultados obtenidos por DeepMind han sido confirmados por los organizadores del experimento sobre las estructuras de las proteínas CASPEnlace externo (ver recuadro), que han calificado la capacidad de cálculo y de predicción de AlphaFold2 como algo “sin precedentes”.
El CASP (Evaluación crítica de la predicción de la estructura de proteínas, por sus siglas en inglés) es un experimento que se lleva a cabo cada dos años y evalúa los avances logrados en el campo de la predicción de la estructura de proteínas a nivel internacional. Durante el último experimento realizado en 2020 (CASP14), se evaluó la precisión de los métodos de predicción en casi un centenar de proteínas diana. Las predicciones de AlphaFold2 demostraron ser muy precisas, incluso en los casos más difíciles, como el de una proteína del SARS-CoV-2 previamente desconocida, la ORF8. El SARS-CoV-2 está compuesto por unas 30 proteínas diferentes, de las cuales una docena no se conoce bien.
La regla de oro del progreso
El eficaz apoyo de la comunidad científica y el intercambio abierto de información en el campo de la biología estructural computacional, al que SWISS-MODEL también ha contribuido, son algunos de los ingredientes que explican el éxito de DeepMind. Según Torsten Schwede, haber compartido abiertamente métodos de cálculo informatizados y datos estructurales ha permitido a DeepMind disponer de la información necesaria para resolver uno de los problemas más difíciles de la bioinformática.
El intercambio de información se intensificó durante la pandemia, lo que demuestra la importancia de la colaboración para lograr resultados significativos en un plazo mucho más breve.»Hemos aprendido mucho durante esta pandemia. El intercambio abierto de información sobre el SARS-CoV-2 dentro de la comunidad científica ha hecho posible el desarrollo de vacunas en un tiempo récord», agrega Schwede, subrayando que almacenar datos de manera aislada puede ser extraordinariamente contraproducente para la ciencia.
Traducción del italiano: José M. Wolff

Mostrar más
Un año Milei; éxito económico en España y la posible producción del VW Golf en México

Mostrar más
La gente joven en Suiza busca empleos con menos riesgo de automatización para la formación profesional

Mostrar más
Suiza y la Unión Europea concluyen negociaciones sobre futuros acuerdos bilaterales

Mostrar más
Siete formas en las que la Convención Europea de Derechos Humanos moldeó la legislación suiza

Mostrar más
España investiga a Airbnb; Trump amenaza con deportaciones y Maduro quiere reformar la constitución




En cumplimiento de los estándares JTI
Mostrar más: SWI swissinfo.ch, certificado por la JTI



Puede encontrar todos nuestros debates aquí y participar en las discusiones.
Si quiere iniciar una conversación sobre un tema planteado en este artículo o quiere informar de errores factuales, envíenos un correo electrónico a spanish@swissinfo.ch.